![]()
![]()
 |
 |
 |
Patient Education
All you wanted to know about perthes disease?
What is perthes disease?
Perthes disease is a disease of the hip in which the bone in the hip joint loses its blood supply. This may cause the bone (femur) to change shape so that it no longer fits into the hip joint (femoral head) properly. The disease run a 2-year course, and if left untreated, can cause permanent damage to the bone.
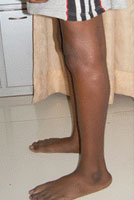
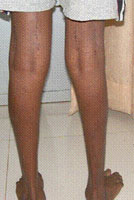
The first stages of perthes disease are silent. This means that the patient dose not limp or complain of pain. The bone in the hip joint (top of the thigh bone or femoral head) loses its blood supply and that part of bone subsequently dies. The bone weakens and a small fracture develops, with the result that the femur head begins to flatten. Circulation returns to the head of the femur within months, rejuvenating the bone but causing pain and a limp (and a visit to the doctor). If left untreated, the femoral head may flatten over the course of months. Leading to arthritis in later years.
What cause perthes disease?
The cause of perthes disease remains unknown. It is, however, an accepted fact that it results from the head of thigh bone. Many reasons have been proposed for this lack of circulation, but none have been confirmed.
Signs and symptoms of perthes disease?
Patients with perthes disease present with a limp, pain, and a stiff hip. When the range of motion at the hip is tested, some movements will be reduced. “Doing the splits” is the first movement in which tightening up is noticed. The thigh often gets thin as well.
Some facts about perthes disease
How is perthes detected?
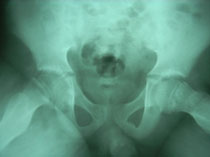
Because many disease processes have signs and symptoms similar to those of perthes disease, it is diagnosed by x-rays. The first x-ray may not be helpful. Because characteristic signs may not have appeared. Bone scans and magnetic resonance images (MRI’s) can be used to detect the amount of blood circulation in the femoral head.
A two-group classification for perthes has been developed. Group A consists hips with less than half of the femoral head involved. This group generally needs little treatment and recovers well. Group B includes hips with more than half of the femoral head involved. Patients in this group need treatment to do well.
What treatment is available for perthes disease?
The aim of treatment is to prevent flattening of the femoral head. There are several methods of treatment: bracing, short periods of rest, soft- tissue releases, and operations on the bone. The choice depends on factors such as the severity of the problem and the stage of the disease.
Treatment is divided into five categories: (1) observation, (2) intermittent symptomatic treatment, (3) definitive early treatment (to prevent deformity), (4) surgery to make the femur head round again, and (5) late surgery for arthritis.
Observation Observation consists of testing the hip stiffness and taking x-ray every few months. It is suitable for children at the time of onset of perthes disease, regardless of the amount of femoral head involvement, provided there is no limitation of movement or subluxation (partial dislocation) of the hip. Observation is also appropriate for children who have Group A perthes with a good range of hip motion and no evidence of femoral head collapse (as indicated by x-rays).
Intermittent symptomatic treatment This treatment consists of rest and exercise when the hip hurts, and may be used in conjunction with observation. It includes muscle-stretching exercise to maintain the joint’s range of motion, and periodic bed rest, with or without traction. X-rays at 3-4 month intervals are necessary during the early months after the first signs and symptoms appear.
Definitive early treatment This type of treatment is used in cases of clinical onset at 6 years of age or older, a Group B hip, and loss of femoral head containment. It involves the use of containment methods (either surgical or non-surgical) early in the disease process.
Containment can be pictured as an ice cream scoop encasing soft ice cream. When the ice cream is enveloped by the scoop (like the head of the femur in the acetabulum or hip socket), the ice cream remains round and undented. Uncontainment occurs when the scoop covers only half of the ice cream. The ice cream outside the scoop dents and the remainder then seeps out. Similarly, the femur is no longer within the acetabulum or socket and a dent develops so the femur is not rounded.
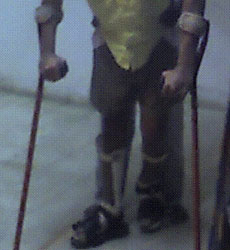
A. Nonsurgical containment (braces and casts) This type of containment is carried out by abducting or drawing away from the body the affected limb in order to place the femoral head back into the acetabulum. The joint is allowed to move, and the movement gradually molds the head into a round shape. Casts and/or braces may be used to keep the femoral head within the joint, and crutches may be used to limit weight bearing on the joint.
Abduction casts consist of long leg casts applied to both lower limbs and held apart by sticks.
A variety of abduction apparatuses and braces are also available. They are lighter, removable, less cumber-some, but also more expensive than casts. Twelve to 18 months of this type of treatment may be required.
B. Surgical containment Surgical containment is indicated when nonsurgical treatment is not feasible or when it is not possible to contain the femoral head in the acetabulum (hip joint) in an abducted position.
A procedure known as an innominate osteotomy is performed. It takes about 2 hours. The purpose of this operation is to use the normal movement of the hip to mold the soft bone into a round shape.
The bones are moved into the desired position and held together with pins. The pins are removed after 6-12 weeks in a minor operation, usually done on an outpatient basis. After surgery the child is often put into a cast or brace until the joint is stable (i.e., the femoral head well contained).
After an 8-week postoperative recovery and a short rehabilitation period, the child is able to resume normal activities. The entire process usually takes about 3-4 months. Making the head round again When there is significant femoral head deformity that cannot be successfully treated with the methods already described, alternative techniques must be sought.
One procedure used is known as a muscle release (of adductors, muscles that draw the limb in toward the centre of the body).this is followed by the use of an abduction cast to hold the limb out. After the patient has worn the cast 3-4 months, enough abduction should be gained so that the femoral head can be placed into the acetabulum through a surgical osteotomy. A second technique involves a combination of femoral and pelvic osteotomies. If the deformed femoral head cannot be contained within the acetabulum, part of the head may have to be removed. Late surgery for arthritis Late surgery is rarely necessary in adults who have had perthes disease as children. Arthritis is usually managed by either an osteotomy to realign the joint or prosthetic replacement of the joint. The method chosen depend on the age of patient.
Prognosis In general, the younger the child at the age of onset of perthes disease, the better the results of treatment. Many long-term studies have shown that most children with clinical onset at less than 6 years of age have an excellent prognosis. However, they must be evaluated through clinical and radiological (x-ray) examinations at intervals of 2 to 4 months. If loss of either motion or containment is noted, a short course (3-6 months) of nonsurgical treatment may be necessary.
The extent of femoral head involvement has much to do with prognosis. Children with less than half of the femoral head involved (Group A) show better treatment results. As loss of containment may still occur, these children must also be examined every 2-4 months. In girls, perthes disease is more likely to involve over half of the femoral head, and so more vigorous treatment is usually needed. Regardless of the method of treatment chosen, it is essential that a good range of motion be achieved for the hip and maintained throughout treatment. The outcome should then be satisfactory.
Tips for parents
Patient Information: Osteogenesis imperfecta (OI)
A newborn lets out a sharp cry while he is being cleaned and weighed. He screams when he is picked up or when someone touches his leg. An x-ray reveals a fractured femur, as well as several healed rib fractures.
Parents bring their one-year-old daughter to the emergency room. She had been pulling to a stand, when suddenly the parents heard a “pop” and the little girl fell to the floor, crying with the pain of a broken leg. This is toddler’s third fracture since birth.
A teenager checks into the hospital for the second time this year. A few months ago, he had a metal rod put into his tibia. This time, he will undergo risky surgery to put a rod in his spine. Doctor hope that the surgery will halt his progressively worsening scoliosis, which is crowding his lungs and leading to repeated respiratory infections. At her annual check-up, a 45-year-old woman asks her physician for a referral to a good orthopedist. The woman had several dozen bone fracture in her childhood and teen years. Though she has been fracture-free for a number of years, she is concerned that menopause will weaken her already fragile bones, leading to another cycle of fractures. The newborn, the toddler, the teenager, and the middle-aged woman all have osteogenesis imperfect, or “brittle bone disorder.” Osteogenesis imperfect (OI) is a genetic disorder that cause fragile bones as well as other health problems. This brochure provides the latest information on osteugenesis imperfecta for health care providers and people affected by OI. What is Osteogenesis imperfecta (OI)? Osteogenesis imperfecta (OI) is a genetic disorder of type 1 collagen—the protein “scaffolding” of bone and other connective tissues. People with OI have a faulty gene that instructs their bodies to make either too little type 1 collagen or poor quality type 1 collagen. The result is bone that break easily.
Four forms of osteogenesis imperfecta have been described, representing wide variation in appearance and severity. It is estimated that between 20.000 and 50,000 people have OI in United States. Below are the clinical features of the four major types of OI. Clinical features vary widely not only between types, but within types, and even within the same family. Many individuals with OI have only some—not all—of the clinical features. Children with milder OI, in particular, may have few obvious clinical features of OI. Type 1
Type 2
Type 3
Type 4
Other features common in people with OI include excessive perspiration, easy bruising, a high-pitched voice, and thin, smooth skin.
Myths and Facts About Osteogenesis Imperfecta
Thanks to the work of dedicated researchers and clinicians. We learn more about osteogenesis imperfecta every day. Most babies born today with OI have a good chance of leading independent, successful, and satisfying lives. Traditional treatments are being perfected, and new treatments for strengthening OI bone are on the horizon. Because OI is a rare disorder, many parents who have a child with OI have never heard of the disorder, and their health care providers may never before have treated anyone with OI. This brochure provides families and medical professionals with updated and accurate information.
Myth: A baby with osteogenesis imperfecta should always be carried on pillow and discourage from moving. Fact: Though there are handling techniques and precautions that are useful when caring for an infant with OI, it is in the child’s best interest to allow him or her to be held and touched, and to explore independent movement to the greatest extent possible. In fact, immobility increase bone loss and fragility, leading to more fractures.
Myth: You can easily distinguish fractures caused by osteogenesis imperfacta from those caused by child abuse. Fact: Children with osteogenesis imperfacta can have all types of fractures—spiral, rib, skull, incomplete, displaced, etc. distinguishing OI from child abuse requires a thorough assessment by a medical professional familiar with OI. Diagnostic test are also available to help confirm a diagnosis of OI.
Myth: Severe cases of OI are often the result of recessive inheritance. Fact: In recent years, OI expert have concluded that recessive inheritance of OI is extremely rare. Most cases of OI are caused by a dominant genetic mutation.
Myth: OI affects only the bones. Fact: Though fragile bones are the hallmark of OI, people with OI often have other medical problems, including loose joints, early hearing loss, brittle teeth, respiration problems, and easy bruising.
Myth: Everyone who has OI is shorter than normal, has blue sclera (whites of the eyes), and uses a wheelchair. Fact: The appearance of people with OI varies considerably. Though many people with OI are short-statured, people with milder forms may be of normal or near-normal height. Some people with OI are born with blue sclera that lighten with age, while others have white sclera from birth. People with OI also have variable mobility, ranging from independent walking to full-time use of a wheelchair.
Myth: Everyone who has OI is diagnosed at birth. Fact: OI type I, the most common and mildest from of OI, is rarely diagnosed at birth. Some very mild cases are only diagnosed when a person has a child with OI type I, and a review of the parent’s medical history reveals that he or she has had fractures and exhibited other features of OI.
How Is OI Inherited? Researchers now agree that nearly all cases of OI are caused by dominant genetic mutation. The majority of children with OI inherit the disorder from a parent. Nevertheless, approximately 25 percent of children with OI are born into families with no history of the disorder. This usually due to a spontaneous dominant mutation that takes place in the sperm or the egg prior to conception.
A person with OI has a 50 percent chance of passing on the disorder to each of his or her children. An affected child will have the same type of OI, as his or her parent. However, the disorder may not affect the child in exactly the same ways it has affected the parent.
When a child with OI is born into a previously unaffected family due to a spontaneous mutation, in most cases the parents do not have an increased risk of having another child with OI. Occasionally, a parent may be mosaic for a mutation; that is, a mutation that causes OI is present in a percentage of the reproductive cells that give rise to his or her sperm or eggs. In this cases, there is a small chance (2 to 4 percent) that the parents will have more than one child with OI. Unaffected siblings of a child with OI are at no greater risk of having children with OI than the general population. How is OI diagnosed? Bone fracture that occurs with little or no trauma are often the first indication that an infant or child may have OI. Babies with Types II,III, and IV are often born with fractures, and/or may show evidence of in utero fractures that have healed. Children with type I often sustain their first fracture(s) as result of normal activity during the first several years of life—during a diaper change, while being lifted or burped, or when they begin standing and walking. Some very mild cases of type I OI are not diagnosed until the teen or adult years.
A physician familiar with OI can often diagnose the condition based on the presence of fracture as well as other clinical features. It is important to remember that the presence of clinical features varies widely among individuals. A positive family history for the disorder can help confirm a diagnosis, although spontaneous mutations do occur in previously families. There are two types of laboratory tests available to help confirm a diagnosis of OI. Neither test is 100 percent accurate, so these test should be considered in conjunction with other factors. A skin biopsy can be analyzed to determine if the quantity or quality of the type 1 collagen is abnormal. A DNA test can be done on a blood sample to try to locate the mutation that caused OI. Approximately 10 to 15 percent of individuals with mild OI who have collagen testing, and approximately 5 percent of those who have genetic testing, test negative for OI despite having the disorder. OI can be prenatally diagnosed in some cases. Ultrasound can often detect bowing, fracture, shortening or other bone abnormalities, particularly in the more severe forms of OI. Type II OI is identifiable by 14 to 16 weeks gestation, and type III OI by 16 to 18 weeks gestation. Even when ultrasound is done by a highly qualified technician or physician, it may not be possible to pinpoint the type of OI (e.g., type II vs. type III). Cells obtained through chorionic villus sampling (CVS) can be analyzed for abnormal collagen as well as for a genetic mutation, while cell obtained through amniocentesis can be analyzed for a genetic mutation. OI is caused by many different genetic testing is generally most useful when the mutation of an affected family member is already known. How is OI Treated? There is no cure for OI. Currently, OI is treated primarily by managing fractures and promoting as much mobility and independence as possible. Prolonged immobility can further weaken bones and lead to muscle loss, weakness, and more fractures. Many orthopedists prefer to treat fractures with short-term immobilization in lightweight casts, splints, or braces to allow some movement as soon as possible after the fracture.
Physical Therapy and Exercise: Physical therapy should begin as soon as it is evident that an infant has muscle weakness or motor skill delay when compared with same-age peers, and continue until a child reaches appropriate physical therapy goals. The long-term goal for children with OI is independence in all life function (e.g., self-care, locomotion, recreation, social interaction, and education), with adaptive devices as needed.
Occupational therapy can help with fine motor skills and adaptive equipment for daily living. As a child with OI grows older and gains more independence, he or she will benefit from continued physical activity, such as adapted physical education. Adults with OI also benefit from safe exercise to maintain bone and muscles mass. Swimming and water therapy are particularly well-suited for people with OI of all ages, as they allow independent movement with little fracture risk. Walking is also excellent exercise for those who are able (with or without mobility aids). Surgery: Many children with OI undergo a surgical procedure known as rodding, in which metal rods are inserted into the long bones to control fractures and improve deformities that interfere with function. There are two basic types of rods. Nonexpandable rods are more versatile but often must be replaced as the child grows. Expandable rods can grow with the bone, but are only appropriate for larger bones (such as the femur) due to their thickness and need to be firmly anchored at both ends. Progressive, sometimes severe, scoliosis is a problem for many people with OI, and may cause respiratory problems. Bracing is generally not recommended, as the force applied may deform the ribs rather than straighten the spine. Spinal rodding may be appropriate in severe cases, if the bone is strong enough to support the rod. Medication and Other Experimental Therapies: Various minerals and medications determine if they strengthen bone in OI. Most of these substances have not been proven effective. Recently, the bisphosphonate drugs (currently approved for use in preventing and treating postmenopausal osteoporosis, and bone complications of certain cancers) have been studied and show some promise for children and adults with OI; research is ongoing. Other treatments, including growth hormone, gene therapies, are also being researched. Healthy Lifestyle: People with OI benefits from a generally healthy lifestyle, including safe exercise and a nutritious diet. Adequate intake of nutrients, such as calcium (to maintain bone density) and vitamin C (to promote healing) is important. However, megadoses of these nutrients are not recommended. Evaluation by a physician or registered dietitian will help people with OI determine adequate nutrient intake for their body size and age. It is also recommended that people with OI avoid smoking, excessive alcohol or caffeine consumption, and steroid medications, which may affect bone density. Are There Precautions to Take When Caring for a Person with OI?
How to choose the perfect shoes for your kid
Patient Information: Choosing Shoes
A shoe is basically a package. But it is a package for delicate object-your foot. Obviously if the package is the wrong shape, or made of wrong materials, your foot can be damaged. So choosing the right shoes is important for anyone, but especially so if you suffer from foot problems.
It might seem odd, but beautiful feet are rarities in western, shoe-wearing societies. In hot countries, where people go without shoes, or wear sandals, ugly or deformed feet are comparatively rare. That says something about the shoes we choose.
Naturally because of our climate and our urban environment we have to wear shoes as protection against the elements, and the right footwear can make a lot of difference. But even if the damage has already been done, or you suffer from arthritis or bunions, care taken now can help make your feet more comfortable. Size of the shoe Selecting the right size of shoes is a basic necessity. The size printed on the shoe, however, is less important than how it feels. Your foot should not be able to slide about too much, but there must be room enough at the front for the natural forward movement of your toes which occurs in motion of walking or running.
Soles of the shoe
These should be light, pliable and hard-wearing. In a wedge-type construction (for instance a shoe with a pointed toe), the sole should be able to bend along a line which could be drawn from the joint at the base of the big toe to the base of the little toe. The best soling materials these days are the moulded or microcellular rubbers which have displaced heavy leather.
Uppers
Leather is still by far the best material for the upper of a shoe. Man-made plastic “leathers” such as you have foot problems. Women with “difficult” feet find that basket-weave uppers are the most comfortable. Both dark colours and a suede finish disguise misshapen feet. You should avoid toe caps. Ornamental stitching, brogue styles and, paradoxically, the most expensive calf leathers all reduce the ability of the upper to mould to the shape of your foot.
Sliding backwards To stop your foot sliding backwards a shoe needs the reinforcement of a heel counter in the heel cup. A sandal requires a sling strap to perform the same function.
Sliding forwards The best way to stop your foot sliding forward is to contain the ”waist” of the foot. Lance-up shoes do this most efficiently; alternatively a Velcro flap or an elasticated panel can do same job. In court-style shoes only the pressure on the toes acts as a buffer.
Toe box This area of the shoe keeps its shape by means of reinforcement built into it. It is also known as the toe puff. The toe box should provide at least one centimeter of room beyond your longest toe.
Checking shoe length Here is a quick and easy way to check if a shoe is the right length for you. Take a strip of flexible card one centimeter wide. Place your foot lengthwise on the strip and mark where your heel and longest toe come.
Cut the strip to this measure and place it inside your shoe (or take it with you when you are buying) with one end of the strip flush to the heel. You should be able to slide the card forward at least one centimeter. Heels Although we were not born to wear shoes with heels, walking is more comfortable with a low heel (about one centimeter) than with none. Women who have always worn high or medium-height heels usually find it difficult to adapt to low heels later on in life.
Slide support Walking shoes, sports shoes and some others have a low, reinforcement wall to stop the foot from sliding sideways. In some sandals or “flip-flop” the same function is performed by a small pillar between the first and second toes.
Children’s shoes Constriction of the toes is the chief cause of deformity in children’s feet. It starts when babies are put into stretch socks and continues if children wear shoes they have outgrown. Adolescent girls (and boys) can rick the health of their feet when they become more fashion conscious and refuse to accept “sensible” footwear.
It is worth encouraging babies and young children to trot around in bare feet or in sandals wherever appropriate. Beyond this, great care should be taken to make sure that all footwear, tights and socks, as well as shoes, fit well and do not compress the feet, especially the toes. Deformities that are acquired before the age of ten may correct themselves naturally as the foot grows. But damage acquired and still remaining up to and during adolescence will be irreversible.
Teenage feet are supple and can often be squeezed painlessly into shoes that are too short or too narrow, but at the same time they are still growing rapidly-often as much as one centimeter in six months. Consequently, a shoe which fits perfectly in January will be either too short or too narrow by July. Bunions (Hallux Valgus) These usually start in childhood and are the direct result of shoes that are too tight. Pointed shoes and court-style are further culprits. The weight of the foot is only kept from sliding forwards by the wedging effect the toe box has on the front of the foot-an X-ray would show this clearly. The toe will inevitably be compressed and, when the shoes are too short or too pointed, bunions are more likely to be caused. These are also more likely if the inner border of the shoe is not straight.
Girls with naturally broad feet face worse problems than normal, since very few fashion shoes are made in more than one or two widths. Damage can, of course, be limited if people wear “sensible” shoes for work and reserve fashion shoes for special occasions. Sports shoes Boys often develop damage because they tend to wear expensive football boots after they have ceased to fit properly. Training shoes have superseded the old gym shoes, which is just as well, since they are better padded and ventilated. Unfortunately, they are seldom manufactured in different widths so, once more, people with broad feet suffer. But it is possible, by shopping around, to find wider shoes for sport
Industrial and safety shoes
Workers on building sites need soles that include a steel plate to protect the foot from the underside. Armoured toe caps are necessary when there is a danger of falling heavy objects. Oil-proof or non-skid compounds may be required if you work in a garage or oily surroundings. If you need rubber boots for outdoor work, you face the same old problem of only one-width fitting. Do not tolerate a boot that is too narrow. Instead, buy a size bigger and pad it with extra socks. Foot problems If you have unusually-shaped feet you can probably be fitted with “depth” shoes often available from chiropodist or the National Health Service.
These have the deeper last to accommodate a moulded insole. You may find that you can get the broader width of shoe you need from a mail order company such as the Bury Boot & Shoe Company, Bury, Lancs. The National Health Service, free of charge, provides custom-made shoes for people with foot deformities, if they can provide a prescription signed by a hospital consultant. This service will vary between areas depending on which shoemaker is contracted to the hospital concerned. A fully hand-crafted pair of shoes could cast the NHS as much as £300! However, proper comfort and satisfaction can usually be found less expensively with “depth” shoes. Also available through the NHS at less cost are shoes made to a plaster- of-Paris replica of the foot in question-constructed from soft leathers bonded to a microcellular sole and padded with modern foam plastics. These are particularly suitable if you are suffering from arthritis in your feet. Advice on where to get these special shoes can be obtained from the local occupational therapy department. Special arthritic problems Early rheumatoid arthritis: Many women manage with sandals in summer and fur-lined boots in winter. Bad circulation, cold feet: Insulated boots such as “moon boots” are helpful.
N.B. There is a danger in wearing these boots for too long as there is no ventilation in them.
Working in the wet or mud: As a substitute for rubber boots, put surgical shoe which inside galoshes or shoe over boots. These are comfortable, warm and waterproof. Stiff ankles: If your ankle is stiff and you can’t point your toes, you will need: (a) exactly the right heel height; (b) shoes which lance right down to toes (these are available in some fashion styles, or, to order, in NHS footwear). Arthritic fingers: If your fingers cannot cope with normal laces, try elastic laces (obtainable from hospital occupational therapy department) or shoes which use a Velcro flap. Otherwise you could buy zip-sided boots and have a split-ring placed in the zip which you can pull up with a hook on a stick. Hallux Rigidus (the big toe is stiff and will not bend upwards): You will need a simple modification to the sole of the shoe so that the foot rocks over the sole instead of the big toe being forced upwards. Chiropody: People with hard skin, corns, in-growing toe-nails should have these attended to before trying shoes on. If chiropody padding is needed, this should be in place before shoe fitting-otherwise the shoe which seems big enough without them be too tight with them. Similarly, if you need shaped insoles taken them along with you to the shoe fitter. Really bad feet (shoes impossible): Hospitals can supply disposable foam plastic “Dru” shoes, which can be padded or cut to fit almost any foot. People with foot ulcers, bulky dressings, protruding toes or awaiting surgery can use these as something to get around in. if soiled they can be washed in a weak solution of bleach and detergent.
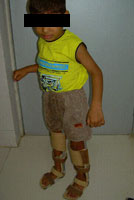
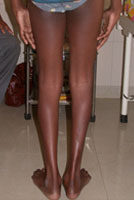
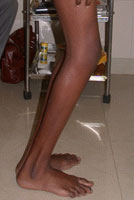
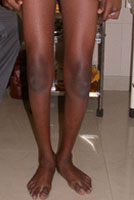
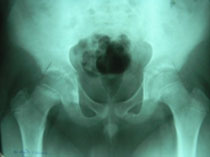
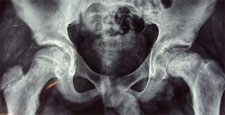
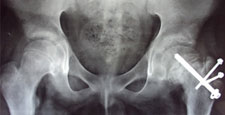
Congenital Dislocation of the Hip (CDH)
What is congenital dislocation of the hip (CDH)?
The hip joint consists of two structures: a cup-shaped socket (acetabulum) in the pelvis and a ball-shaped upper end of the thigh bone (femoral head). Normally, these structures fit snugly together and are held in place by ligaments and muscles. In congenital dislocation of the hip (CDH), the femur comes out of the acetabulum. (See Glossary at end for definitions of technical terms.)
There are two categories of CDH, typical, which occurs at birth, and teratologic, which occurs prenatally. If the dislocation is left untreated, permanent problems such as a limp, a short leg, and a diminished range of movement may result.
What causes CDH? The most common cause of CDH is looseness (laxity) of the ligaments surrounding the hip. This is likely due to fetal absorption of the maternal hormones that increase pelvic relaxation just before delivery uterine compression, which forces the infant’s hips into a flexed position and limits fetal movement, is also thought to play a role in CDH.
Sometimes the way the newborn child is held or is placed in the crib may contribute to the development of hip instability and subsequent dislocation.
Some facts about CDH
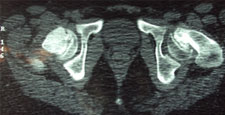
Diagnosis of CDH: After the neonatal period
By the time the infant is 6 weeks of age, the Ortolani and Barlow test are no longer effective diagnostic tools because the soft tissues around the hip joint have tightened.
Diagnosis of CDH: Age 6 weeks to one year As the infant grows the motion of the affected hip will become restricted. That is, abduction will be limited. An x-ray is required to make a diagnosis at this stage.
Diagnosis of CDH: Age 1 year If the child is not treated before the age of 1 year, he or she will walk with a limp and leg abduction (doing the splits) will be restricted. The affected limb will also be shorter than the other. Although the child may appear disabled, pain is uncommon and limitations on leg function are slight. Once again, an x-ray is required to make the diagnosis.
Diagnosis of CDH: Adult The untreated adult will walk with a limp, have legs of unequal length, will a limp, have legs unequal length, will experience fatigue when walking, and may feel pain. Again, an x-ray is useful for diagnosis.
Where can advice be obtained? If your physician detects hip instability in your infant that does not resolve itself with in 5 days od detection, a referral should be made to an orthopaedic surgeon. The surgeon will check the findings, have an ultrasound test done, make a diagnosis, and begin treatment if it is required.
Treatment for CDH The goal of treatment is to place the femoral head back into the acetabulum and keep it there until the structures surrounding the hip (muscles, ligaments, and tendons) become strong enough to hold joint together.
Method of treatment and results vary with the age of the child at the time of diagnosis. Generally, the younger the patient when diagnosed, the better prognosis. Both complexity of treatment and risk of complication tend to increase with age. Treatment for CDH: Age 0 to 6 month For typical CDH, treatment consists of bracing and splinting. Examples of equipment used include plastic-covered metal splints, abduction pillows, and cloth harnesses (N.B. triple diapers are not a reliable treatment method). Occasionally a spica cast may be used. A common treatment choice at this stage is the pavlik harness. Patients usually wear their treatment apparatus continuously for 2 to 4 months. After this period, a night splint may be used for 2 to 6 months to ensure that healing is complete. Treatment complication are rare for those with typical CDH in this age group.
About 15% of patients are not successfully treated with the harness and may require traction followed by a cast. A few require an operation. For those with teratologic dislocation (a rare kind of stiff dislocation present before birth), treatment is more difficult. Muscle releases, traction, and open reduction (surgery) is often required. Surgery usually takes about 2 hours but varies with the complexity of the situation. The prognosis for normal joint development is poor.
Treatment for CDH: Age 8 to 18 months Treatment of the older infant with previously undetected CDH is more difficult. Closed reduction (putting the hip back into the joint and holding it with a cast) can be attempted after a preliminary period of traction to stretch the tissues around the hip. Home traction may be required for 10 days to 3 weeks.
Treatment for CDH: Age 18 month to 4 years
Surgical treatment is almost always necessary for children diagnosed during this period. A combination of treatments may be required to achieve hip stability. These include traction, muscle release, open reduction, and innominate osteotomy (followed by a 6-week period of casting). Treatment at this stage is technically demanding, but the prognosis for long-term function is good.
Open reduction (surgery) may be required if closed reduction is not easily achieved. After reduction, the child is immobilized in a series of casts. From 4 to 8 months of cast treatment may be required for additional 4 to 6 months. The majority of children treated with closed reduction and traction develop normal hips. In 10% of the children, however, avascular necrosis occurs (death of bone due to loss of blood supply). Complications of treatment
Tips for parents of CDH Patients
Myotonic Dystrophy
WHAT IS MYOTONIC DYSTROPHY?
Myotonic dystrophy, also known as Steinerts’s disease, is the most frequently diagnosed adult form of muscular dystrophy. It is characterized primarily by progressive muscle weakness and muscle wasting and by myotonia (difficulty in relaxing a muscle or a group of muscles following contraction). It is a multisystem disease, typically involving a wide range of other tissues as well as muscle.
WHO CAN BE AFFECTED BY MYOTONIC DYSTROPHY?
Myotonic dystrophy is a hereditary disorder transmitted from generation to generation by a parent who has the disorder. Myotonic dystrophy can affect either males or females.
Approximately one person in 8,000 around the world is affected by myotonic dystrophy, but it occurs more frequently in some populations than in other. For example, in northern Quebec the incidence of myotonic dystrophy is exceptionally high, with up to one person in 500 being affected. AT WHAT AGE CAN A PERSON BE AFFECTED? Myotonic dystrophy can occur at any age, though a high percentage of affected people are diagnosed by their early twenties. An unusual feature of myotonic dystrophy is known as anticipation. Anticipation refers to the tendency for myotonic dystrophy to become more severe and the age at which symptoms appear to occur earlier as it passes down the generations of a family.
Congenital myotonic dystrophy is the most severe form of myotonic dystrophy. Symptoms of this form of myotonic dystrophy are always evident at birth. In fact, decreased movements may be noted in the unborn child as early as the second trimester of pregnancy. Virtually all infants with congenital myotonic dystrophy are born to mothers who also have myotonic dystrophy. IS MYOTONIC DYSTROPHY ANYONE’S FAULT? No. myotonic dystrophy is a genetic disorder that is passed from one generation to next. It is not anyone’s fault.
IS IT CONTAGIOUS? No. genetic diseases are not contagious.
WHAT CAUSES MYOTONIC DYSTROPHY? Myotonic dystrophy is caused by a mutation in a gene located on the log arm of chromosome 19.
In 1992, the gene responsible by Dr. Robert Korneluk and his Canadian research team, in collaboration with groups in the Netherland, California and Britain. These distinguished scientists are continuing their research to determine exactly how, when faulty, it causes myotonic dystrophy.
HOW DO GENE ABNORMALITIES CAUSE MYOTONIC DYSTROPHY? The basic fault is an unstable inherited mutation in a particular gene known as the myotonic dystrophy protein kinase gene. At the present time, it is not known how the genetic abnormality causes the disorder.
WHAT ARE GENES? Genes are basic function units of heredity that tell the cells and tissues of the body what specialized functions they will perform. They are found in the nucleus of the cell as tiny segments of DNA in the chromosome.
There are approximately 80,000 individual genes located on the 46 chromosomes (23 pairs) inside each of the billons of cells within the human body.
HOW DO GENES FUNCTION? Genes are the blueprints for structure and function. Each gene carries the code that directs the cell to make a specific protein. Proteins are the molecules that carry out all of the work of the living body. The proteins coded by the genes are responsible for all structures and functions of living cells from eye colour to muscle function. They can also be hormones or enzymes within the body.
WHERE DO THE FAULTY GENES COME FROM? When a baby is formed, he/she receives 23 chromosomes from each parent, for total of 46 chromosomes (23 pairs). Normally, each pair of chromosomes carries the same genes for the same traits. 22 pairs of the chromosomes are called autosomal chromosomes, which simply means that they are identical in both males and females. The chromosomes in the 23rd pair are known as the sex chromosomes and it is here that the sex of the unborn child is determined. Genes are found packed together on these chromosomes. Each gene has a precise location on one of them. For reasons that are only partly understood, one or more genes may become flawed or lost, and a serious disorder may result.
Depending on the type of neuromuscular disorder, the faulty or missing gene may be inherited. Also, the way that the disorder is inherited will vary from disorder to disorder. HOW IS MYOTONIC DYSTROPHY INHERITED? Myotonic dystrophy is transmitted through autosomal dominant inheritance. Autosomal refers to the fact that the faulty gene can appear on any of the 22 chromosome pairs not associated with determining the sex of the child. In case of myotonic dystrophy, the faulty gene is found on chromosome 19.
Dominant refers to fact that the disorder is passed down from a parent of either sex who has the disorder. With each pregnancy, there is a 50% risk that the child will receive the faulty gene from the affected parent and have myotonic dystrophy. There is a 50% chance that he/she will receive normal versions of the gene from each parent and will therefore be unaffected. WHAT ARE THE DIFFERENT FORMS OF MYOTONIC DYSTROPHY? There are forms of myotonic dystrophy. They are the adult form and the congenital form.
Adult Form This form generally becomes apparent when a person is between 10 and 30 years old. But in some cases, symptoms may appear in younger children or much later in life.
The course of myotonic dystrophy varies widely form one person to the next, even among members of the same family. Some people with the disorder have such mild symptoms that they do not suspect anything wrong and adapt very easily. Others may be faced with a significant level of disability, requiring assistance with mobility and activities of daily living. In the adult form of myotonic dystrophy, muscle weakness usually starts gradually and progresses slowly. People with this form often mention stiffness as an early symptom. Disability typically occurs 15 to 20 years after the onset of the disease when the weakness affect the shoulder muscles, thighs and hips, though many people with myotonic dystrophy live their entries lives without any detectable symptoms. Congenital form The congenital form of myotonic dystrophy, as its name implies, is always present at birth. Affected infants are profoundly weak and have many difficulties. Frequently, there are problems associated with breathing, sucking and feeding.
The babies are extremely hypotonic (decreased muscle tone) at birth, and as a result they appear floppy. Some babies die within a few hours or days of birth. Infants with congenital myotonic dystrophy have weakness in virtually all muscles, including the respiratory and facial muscles. If the infant survives the newborn period, a gradual improvement is generally noted in his/her respiratory function and ability to swallow. As they grow older, children are slow in developing language quently in these children, making it difficult or impossible for them to live totally independent lives as adults. HOW ARE MUSCLES AFFECTED? In the adult form of myotonic dystrophy, muscle weakness is reported by over 60% of clients. It is the most common symptom resulting in referral to a physician. Hands and feet are normally the first parts of the body to be affected. Weakness may then spread gradually into the muscles of face, neck, arms and legs.
The following signs of muscles weakness may be present in affected individuals:
The Hair
Quite often, premature baldness may occur on the forehead and top of the head in males and women may experience abnormal thinning of their hair.
WHAT OTHER SYPMTOMS MAY BE EXPERIENCED? Sleep and personality: Many people with myotonic dystrophy tend to fall asleep easily, sleep for long periods of time and feel excessively drowsy while awake. This characteristic is known as somnolence. It may be so noticeable that it affects an individual’s ability to hold a job. Individuals affected by myotonic dystrophy may be very easy going or at times they may be listless. They are often content to remain inactive for long periods of time. Work: As a person’s myotonic dystrophy progresses, the individual may experience difficulty in performing manual tasks or maintaining a good pace at work. Reorganizing or restructuring responsibilities on the job may help the person to remain active and productive for a longer time.
Reaction to Drugs: People with myotonic dystrophy may react in unusual ways to certain drugs. It is better to refrain from taking any non-prescription drug without the advice of a physician. Some people with myotonic dystrophy have an increased sensitivity to anesthetic agents and other muscle relaxants. Administration of these drugs might results in excessive drowsiness or other side effects. This sensitivity has been noted both in clients who are severely affected by myotonic dystrophy and in those whose symptoms are mild.
HOW IS MYOTONIC DYSTROPHY DIAGNOSED? A diagnosis of myotonic dystrophy is made by a physician. A physical examination and a complete history are necessary for a doctor to assess the presence of myotonia, muscle weakness and any decrease in muscle size. Details of a positive family history, if present, will be investigated, as well as the history of the client’s present concerns.
Diagnostic tests are also used by the physician in making a positive diagnosis of myotonic dystrophy. WHAT DIAGNOSTIC TESTS ARE PERFORMED?
WHERE CAN ADVICE BE OBTAINED?
Neurologists, doctors who specialize in disorders affecting the nervous system, are often consulted for patients who may have myotonic dystrophy. Nerves and muscles are part of the so-called peripheral nervous system.
Neuromuscular clinics and hospital across Canada are equipped to do the necessary diagnostic testing and to offer advice and support regarding the ongoing management of myotonic dystrophy.
Geneticists, doctors who specialize in medical genetics, molecular geneticists and genetic counselors are employed by major hospitals across Canada and are available to patients and their families for the purpose of diagnosis and counselling. A geneticists or a genetic counselor will be able to advise family members about their risk of having an affected child in families where there is a positive history of myotonic dystrophy.
IS THERE A TREATMENT OR CURE AVAILABLE FOR MYOTONIC DYSTROPHY? At the present time, there is no cure or treatment available that can prevent or delay the progression of muscle weakness caused by myotonic dystrophy.
Staying as active as possible remains the most effective way of maintaining the best physical condition. Rehabilitation specialists (physiotherapist and occupational therapists) can advise individuals affected by myotonic dystrophy and their families on how to create a stimulating environment. Also, they can prescribe an exercise program and aids that can assist an individual to carry out activities of daily living. There are medications available that can decrease the effects of myotonia, but they sometimes cause undesirable side effects. Drugs are usually prescribed only when myotonia prevents a person from working or performing daily activities. When necessary, surgery is available to clients who have cataracts. The operation, that may require a short stay in hospital, can result in substantially improved vision. Speech therapy can provide assistance to a child or adult who has difficulty speaking. An adapted educational environment can promote the development of a child’s abilities within school setting. Arrhythmias, dizziness and other symptoms associated with heart problems may be treated with medication when they are causing difficulties. If cold lasts for more than two weeks, treatment may be indicated. As well, some people may find it useful to be vaccinated against the flu
Digestive problems may be decreased by attention to healthy eating habits. Some general suggestions include:
If problems with constipation or stomach irritation persist, medical treatment based on the individual’s complaints may be necessary.
GENERAL INFROMATION FOR THE PERSON WITH MYOTONIC DYSTROPHY
WHO ARE THE MEMBERS THE HEALTH TEAM?
A family will be seen by their own family doctor and possibly, other health professionals. If necessary, they will be referred to a neuromuscular clinic as near to their home as possible.
Treatment, if any, will vary according to the severity of weakness and the related problems that a person is experiencing. Because the course of myotonic dystrophy is so variable, even among members of the same family, it is difficult to be specific about who will need to be consulted. The following information is intended to serve as a guide to medical professionals with whom you may be in contact. The physician (PT) will teach chest physiotherapy techniques to children and adults that will enhance their physical wellness. The physiotherapist helps clients develop individual exercise programs to maintain optimum muscle strength. This program will include the and amount of activity that is best for each person. The PT may also be consulted should respiratory difficulties occur. The geneticist will help individuals and their families to understand how this disorder has been transmitted within their family, as well as the impact of myotonic dystrophy on future generations. The occupational therapist (OT) can help clients learn how to adapt to physical limitation. The OT will be involved, as required, in choosing equipment and to enhance function at school or in the workplace. In conjunction with an infant development worker, the OT/PT may have suggestions for activities to stimulate intellectual development of babies and children with myotonic dystrophy. The role of the nurse, whether in the clinic or the community, is to work with the family to understand the disorder and its management. The social worker, in conjunction with the nurse, provides support to the individual and family member coping with the effects of myotonic dystrophy.
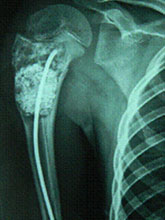
Unicameral Bone Cyst (UBC) or Simple Bone Cyst (SBC)
Unicameral Bone Cyst (UBC) or Simple Bone Cyst (SBC) constitutes 3 % of all bone tumours.
Cyst that Require treatment
Treatment Modalities
About Bone Cyst Bone Cyst is a curable condition. Spontaneous resolution after a fracture is known. Success rate with Intra-Lesional Steriod and Bone Marrow is about 80%. Currettage, bone grafting and intra-lesional rodding with titanium implants or screws have over 90 % success rate in curing the lesion.
Surgery of Cerebral Palsy
Single Event Multilevel Surgery (SEMLS) & Percutaneous Apneurotic Slide Surgery (PERCS)
Most children with Cerebral diplegia with develop muscle tightness and imbalance during period of rapid growth spurts. This can make standing and ambulation difficult. The muscle tightness and spasticity also consume energy leading to early fatigue and reluctance for the child to stand or walk.
Staged surgery and repeated plaster cast treatment cause muscle wasting and weakness.
The concept of SEMLS and PERCS were developed with the common goal to achieve the most optimal results with minimal immobilization.
The surgery is performed under regional anesthesia and post-operative rehabilitation commences as soon as the child is comfortable.
Percutaneous aponeurotic slide surgery (PERCS)
Crouch gait correction involves muscle rebalancing procedures and require dedicated post operative physiotherapy to achieve optimal results. About Cerebral Palsy Surgery Management of children with cerebral palsy need an integrated approach. A systematic assessment is carried out prior to surgery to identify the goals of SEMLS or PERCS for that particular child and to determine what is called the “Surgical Dose” required to achieve optimum results.
The surgery is planned after thorough discussion with the parents /caregivers and the treating physiotherapists so that the post operative rehabilitation protocol is tailored to the individual child. Appropriate orthotics are also fabricated so that plaster immobilization is minimal to prevent muscle disuse. The child is also prescribed appropriate analgesics and muscle relaxants to ease the pain and spasm which can sometimes occur in the post-operative period.
Occasionally Botulinum toxin injections are combined with surgery to decrease the need for extensive muscle releases. Please don’t hesitate to ask your doctor or physiotherapist whether SEMLS or PERCS is appropriate for your child.
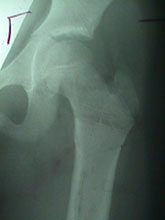
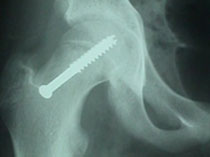
Slipped Capital Femroal Epiphysis (SCFE)
SCFE is a common hip pathology seen more frequently in boys than girls during the adolescent growth spurt. There is a mechanical weakening between the met- aphyses of the femoral neck and head causing the epiphysis to slip posteriorly.
It is more common in children with endocrine disorders, with metabolic bone disease and renal disorders. Children with bilateral slips must be evaluated for thyroid and, growth hormone deficiency and renal causes SCFE may vary from mild (early) slip to moderate and complete slip depending on the extent of displacement of epiphysis. SCFE can also be classified as Stable and Unstable slips depending on the ability of the child to bear weight on the affected leg.
Any acute, and unstable SCFE requires emergent treatment. Stable slips also need to pinned on an elective basis. Untreated or unreduced SCFE can lead to Avascular Necrosis of the epiphysis, chondrolysis and Femoral-Acetabular impingment leading to early hip arthritis. Severe Slip may required open reduction using the Anterior or Watson-Jones Approach or the Safe-Surgical Hip (GANZ) Dislocation approach.
SCFE treated with the Ganz Approach
Torticollis or Wry Neck Deformity in Children
Common in the first decade
In the newborn may present as a small swelling in the substance of the sternocleidomastoid muscle
Clinical Features
Treatment Modalities
About Torticollis This is a common soft tissue problem in neonates and may co-exist with developmental dysplasia of the hip joint (DDH) and calcaneovalgus foot. Over 90% may resolve spontaneously by six to 12 months. 10 % cases need physiotherapy and about 5 % of these may progress to a rigid deformity.
Physiotherapy and local injection of Botulinum toxin can help in the early stages of spasmodic torticollis and some cases of muscular torticollis. Once the deformity is established, surgery is usually recommended by four years of age to prevent or reverse the secondary associated features such as facial hypoplasia and improve the visual field.
Surgery is safe and always performed under anesthesia with muscle relaxation. The technique involves a special type of muscle lengthening which you can discuss with your doctor. The child has to use a special differential collar for 6—12 weeks post-operatively.
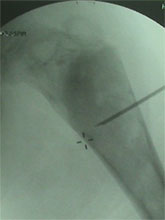
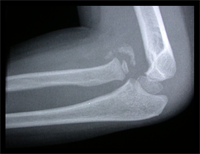
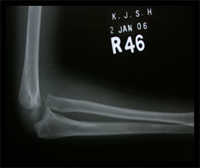
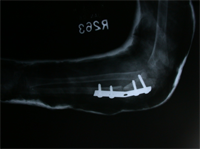
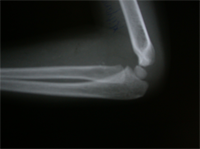
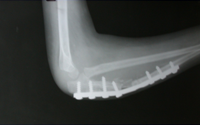
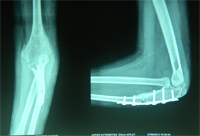
Monteigga Fracture – Dislocation.
The Monteigga Injury
The Monteigga-fracture dislocation is an injury complex where there is fracture of the ulna and dislocation of the radial head. The ulnar fracture may be incomplete or complete and sometimes there is only plastic deformation of the ulna bone. The injury assumes importance due to the frequency with which the radial head dislocation is missed. Acute injuries of the forearm should always be followed by thorough examination of the wrist and elbow.
Biplanar radiographs should include the wrist joint and elbow joint to rule out any joint subluxation or dislocation. Two cases of late presenting Monteigga Fracture — Dislocation. Clinical Features
Treatment Options
The main aim of the operation is to achieve a stable radial head reduction without significant loss of joint motion
Open reduction of the radio-capitellar joint with ulnar angulation osteotomy. The ulnar—distraction osteotomy to achieve radial head reduction. No ligament reconstruction was required. Monteigga Fracture – Dislocation.
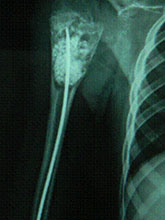
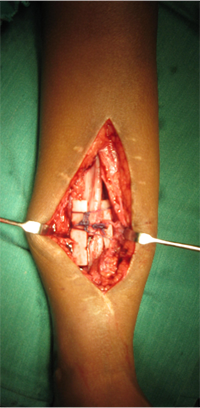
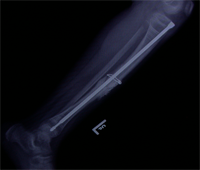
Congenital Pseudarthrosis of Tibia
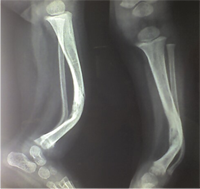
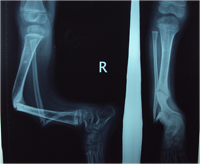
Congenital Pseudarthrosis of the Tibia
Also known as congenital "Anterolateral Bowing of Tibia" or CPT is a rare condition seen in infants and children. The tibia is bowed and the apex of the deformity in anterior and lateral in the sagittal and coronal plane respectively. Incidence is 1: 140,000. This type of bowing is pathognomomic of CPT or Congenital Pseudarthrosis of tibia, leading to a recalcitrant non-union of the tiba and fibula. Almost 50% of cases are associated with Neurofibromatosis type I or fibrous dysplasia. The deformity may be present at birth or occurs as a painless stress fracture when child starts to walk. The typical site of deformity is in the lower third of the leg and both tibia and fibula may be involved. Radiographs reveal typical bowing and the tibia may have sclerotic edges, cortical tapering, cyst formation and sometimes obliteration of meduallary cavity. CPT that Require Treatment
Treatment Modalities
Prognosis doesn't depend on the type of CPT but the extent of involvement of the bone, presence of gap or stiff non-union, and the age of patient at treatment.
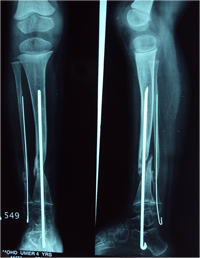
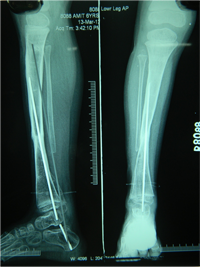
Rush Rodding Periosteal Grafting
Newer Fassier - Duval rod in an older child. About CPTCPT is a disorder of the tibia where there is a propensity for recurrent fractures, poor healing of bone and need for multiple operations. Current methods of treatment of CPT have evolved to give much more consistent results than previously possible. Special methods are available to achieve union. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||